
New Drug Candidates in AACR2023 (“New Drugs on the Horizon”)
작성자
관리자
작성일
2023-04-19 14:40
조회
1884
2023년 4월 16~19일, 미국 올랜도에서 개최된 2023년 미국암연구협회(AACR, American Association of Cancer Research) 연례학회에서 처음 구조와 함께 연구결과가 공개된 저분자 혁신신약 후보물질 10종 (PROTAC 3종, ADC 2종 포함)을 소개합니다. (“New Drugs on the Horizon”)

 Discovery of RMC-6291, a tri-complex KRAS-G12C(ON) inhibitor
Discovery of RMC-6291, a tri-complex KRAS-G12C(ON) inhibitor
대부분의 기존 KRAS 저해제는 inactive, GDP-bound form of KRAS와 결합하여 작용하는 반면, ‘RMC-6291’은 activated, GTP-bound forms of KRAS와 공유결합을 통해서 비가역적으로(irreversible) 지속적으로(durable) KRAS-G12C-mutated tumors에 효과를 나타낸다.
AACR2023 Abstract (바로가기)
The KRAS-G12C mutation occurs in 11 - 14% of non-small cell lung cancers, ~4% of colorectal cancers, and ~2% of pancreatic cancers in the U.S., and drives these cancers by shifting the cellular equilibrium of KRAS towards the GTP-bound, active state, KRAS-G12C(ON). The resulting increased levels of KRAS-G12C(ON) in turn increase signaling output to initiate and support the oncogenic state. RMC-6291 is a potent, orally bioavailable inhibitor of KRAS-G12C(ON) that forms a tri-complex within tumor cells along with KRAS-G12C(ON) and cyclophilin A (CypA), driving a near-immediate disruption of RAS effector binding and extinction of KRAS-G12C(ON) signaling. RMC-6291 treatment produces deep and durable suppression of RAS pathway activity in KRAS-G12C tumor models and drives profound tumor regressions in vivo. In a mouse clinical trial with a representative panel of xenograft models of KRAS-G12C NSCLC, RMC-6291 outperformed adagrasib, displaying a potential ‘best-in-class’ profile with an increased number of responses, greater depth of tumor regressions and improved durability of responses. Thus, RMC-6291 is a clinical stage inhibitor of KRAS-G12C(ON) that potentially overcomes limitations of first-generation KRAS-G12C(OFF) inhibitors.

 Discovery of ARV-766, an androgen receptor degrading PROTAC® for the treatment of men with metastatic castration resistant prostate cancer (mCRPC)
Discovery of ARV-766, an androgen receptor degrading PROTAC® for the treatment of men with metastatic castration resistant prostate cancer (mCRPC)
AACR2023 Abstract (바로가기)
Prostate cancer is the second leading cause of cancer death in men in the United States. The androgen receptor (AR) plays critical roles in both early disease and advanced prostate cancer. Current therapeutic approaches targeting the androgen/AR axis are initially effective, but castration resistant prostate cancer (CRPC) inevitably develops. CRPC is linked with increased AR activity via gene overexpression, amplification, and gain-of-function mutations. To address these mechanisms of AR-dependent prostate tumor growth, we have developed a novel therapeutic agent, ARV-766, a proteolysis targeting chimera (PROTAC®) that induces a protein-protein interaction between the AR and specific E3 ubiquitin ligase complexes, leading to the ubiquitination of AR and its subsequent degradation via the proteasome. In vitro, ARV-766 degrades AR in various prostate cancer cell lines, including those harboring resistance-conferring, clinically relevant point mutations, with a half-maximal degradation concentration (DC50) of <1 nM in wild type VCaP. Importantly ARV-766 also maintains potency against the AR L702H mutant, which has been associated with resistance to some AR antagonists. In vivo, ARV-766 is orally bioavailable and robustly degrades AR with a >90% observed maximum degradation (Dmax) at efficacious doses. ARV-766 significantly and dose-dependently inhibits tumor growth in murine LNCaP and VCaP xenograft models, including an enzalutamide-insensitive non-castrated VCaP model. These preclinical data supported the clinical development of ARV-766 for the treatment of men with metastatic CRPC. Selected pre-clinical data along with the chemical structure of ARV-766 will be presented.
In the New Drugs on the Horizon session at AACR, Bayer presented preclinical data on its novel selective diacylglycerol kinase (DGK) zeta inhibitor BAY 2965501, which is under Phase 1 clinical evaluation. The enzyme DGKzeta is a lipid kinase that can down-modulate T-cell activation by catalyzing the conversion of diacylglycerol to phosphatidic acid, thus acting as a ligand-independent, intracellular immune checkpoint. An inhibition of DGKzeta has been demonstrated to enhance T-cell priming against suboptimal tumor antigens and has the potential to overcome multiple immune-suppressive mechanisms in the tumor microenvironment. BAY-2965501 is under development for the treatment of skin cancer, kidney cancer, stomach cancer, solid tumor, gastroesophageal junction adenocarcinoma and non-small cell lung cancer. The therapeutic candidate administered through oral route and acts by targeting diacylglycerol kinase zeta (DGKZ).
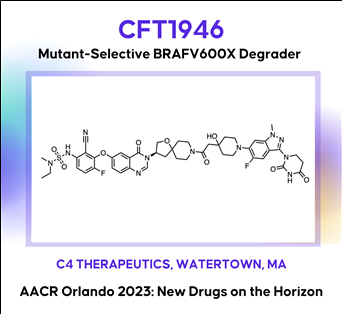
 The discovery and characterization of CFT1946: A potent, selective, and orally bioavailable degrader of mutant BRAF for the treatment of BRAF-driven cancers
The discovery and characterization of CFT1946: A potent, selective, and orally bioavailable degrader of mutant BRAF for the treatment of BRAF-driven cancers
AACR2023 Abstract (바로가기)
The BRAF kinase plays a critical role in the MAPK signaling pathway and is mutated in ~8% of all human cancers including melanoma (~60%), thyroid (~60%), and lung adenocarcinoma (~10%). The most common mutation in BRAF is V600E (Class I), which is found in >70% in these cancers. Despite the clinical success of approved small molecule inhibitors of BRAF V600E (vemurafenib, dabrafenib and encorafenib), this remains an area of unmet medical need because nearly all patients progress, due to either primary or acquired resistance. A bifunctional degradation activating compound (or BiDACTM degrader) may address the liabilities of approved drugs by overcoming, or preventing the emergence of, resistance to BRAF inhibitors. Here we describe CFT1946, an orally bioavailable cereblon-based BiDAC degrader of BRAF V600 mutant proteins, and provide an overview of the medicinal chemistry path leading to its discovery. CFT1946 degrades BRAF V600 mutant proteins, while maintaining exquisite selectivity against the proteome, sparing wild type BRAF (BRAF-WT), ARAF, and CRAF. In A375 cells, CFT1946 potently degraded BRAF V600E and inhibited ERK phosphorylation and cell growth while having no effect in the mutant KRAS, BRAF-WT driven cell line HCT116. In the A375 xenograft model, oral delivery of CFT1946 at 10 mg/kg PO BID resulted in deeper and more durable tumor regression compared to a clinically relevant dose of encorafenib. Further evaluation of CFT1946 in an engineered, clinically relevant BRAFi-resistant A375 cell line (endogenous BRAF V600E + engineered expression of NRAS Q61K) demonstrated that CFT1946 both degraded BRAF V600E and caused a loss of viability in these cells, while treatment with encorafenib had no effect. In xenografts derived from this BRAFi-resistant cell line, oral dosing of CFT1946 as a single agent led to tumor growth inhibition, while treatment with a clinically relevant dose of encorafenib had no effect on tumor growth. Furthermore, dosing CFT1946 in combination with the MEK inhibitor, trametinib, resulted in significant tumor regression, whereas combining encorafenib with the same dose of trametinib had no effect. The medicinal chemistry campaign resulting in CFT1946 focused on the improvement of in vivo pharmacokinetics and rational linker design to achieve high oral bioavailability in a beyond Rule of 5 heterobifunctional degrader. The preclinical data presented herein support the planned Phase 1/2 clinical trial of CFT1946 for the treatment of BRAF-V600 mutant solid tumors.
AACR2023 Abstract (바로가기)
Although the treatment landscape of advanced urothelial cancer (UC) has changed dynamically over the last decade, metastatic UC still has amongst the worst 5-year recurrence rate of any cancer type, highlighting the need for new therapeutic options for these patients. Analogous to breast cancer, advanced UC is a heterogeneous disease, with luminal and basal subtypes. For luminal breast cancer, small molecule targeting of the lineage-defining transcription factor estrogen receptor (ER) is a demonstrated and effective therapeutic strategy. Two-thirds of advanced UC is classified as luminal, which is characterized by overexpression of the urothelial lineage-defining transcription factor PPARG. Thus, small molecule targeting of PPARG in luminal UC may serve as a therapeutic strategy analogous to ER in luminal breast cancer. While PPARG agonists have been well studied in the context of metabolic disease, PPARG inhibitors have received far less attention. Previously reported PPARG inhibitors display minimal phenotypic activity in pre-clinical UC models, calling into question the performance of these molecules, or alternatively, the role of PPARG as a survival lineage oncogene in UC. Leveraging reconstituted, multi-component PPARG biochemical systems, we first identified the limitations of previous tool compounds and then discovered a novel, covalent lead series with unique, context-dependent electrophilic properties. These efforts lead to the discovery of FX-909, a first-in-class covalent PPARG inverse agonist with powerful repressive conformational biasing activity. FX-909 is highly potent in cells (IC50=1 nM), demonstrates high specificity for PPARG (>2000-fold selective over PPARA/PPARD), and elicits robust in vitro growth inhibition in UC cell lines with activated PPARG signaling. Tumor regression was observed in UMUC9 (PPARG amplification) and HT1197 (RXRAS427F hotspot mutation) xenograft models of UC with oral dosing at 1 mg/kg. Predictable, on-target and reversible pharmacology was observed at FX-909 doses above 1 mg/kg, mimicking PPARG loss-of-function mutations with notable tissue remodeling in adipose tissue and the normal urothelium. These collective findings corroborate the role of PPARG as a key UC survival oncogene and suggest that FX-909 will be an effective therapy for patients with advanced UC harboring the luminal subtype.


Preclinical development of ABBV-319: a CD19-targeting glucocorticoid receptor modulator (GRM) agonist antibody-drug conjugate (ADC) for the treatment of B-cell malignancies
AACR2023 Abstract (초록바로가기)
Introduction: Glucocorticoids are key components of standard-of-care treatment regimens (e.g., R-CHOP, Hyper-CVAD) for several B-cell malignancies. However, prolonged systemic glucocorticoid treatment results in glucocorticoid-associated adverse events and acquired resistance that limit its therapeutic potential. ABBV-319 is a novel CD19-targeting ADC engineered to reduce glucocorticoid-associated toxicity observed with systemic glucocorticoids while possessing three distinct mechanisms of action (MoA) to increase efficacy: 1) antibody-mediated delivery of GRM to activate glucocorticoid receptor (GR) induced cell death in cancer cells, 2) inhibition of CD19 signaling, and 3) enhanced Fc-mediated cancer cell killing via afucosylation of the antibody backbone.
Results: We identified a GRM agonist that is 15 and 150 times more potent at driving GR transcriptional activation and cell death compared to clinical glucocorticoids dexamethasone and prednisolone, respectively. The conjugation of GRM agonist as the payload on ABBV-319 enables potent GRM-driven anti-cancer activity against malignant B-cell lines in vitro as well as in cell-line and patient derived xenograft (CDX and PDX) models in vivo. Remarkably, a single-dose of ABBV-319 induced sustained tumor regression and enhanced anti-tumor activity compared to repeat dosing of systemic glucocorticoids (e.g., prednisolone) at its maximum tolerated dose in mice. The CD19 monoclonal antibody (mAb) also reduced proliferation of a subset of B-cell malignant cell lines through inhibition of the PI3K/AKT pathway. Moreover, afucosylation of the CD19 mAb in ABBV-319 enhanced Fc-mediated antibody-dependent cellular cytotoxicity (ADCC), and this activity was maintained after conjugation with GRM payloads. ABBV-319 bound similarly to both V158 (high-affinity) and F158 (low-affinity) FcγRIIIa allotypes and mediated potent ADCC in co-culture assays with human peripheral blood mononuclear cells (PBMCs). Notably, ABBV-319 displayed superior efficacy compared to afucosylated CD19 mAb in human CD34+ hematopoietic stem cell-engrafted NSG-Tg(Hu-IL15) mice, demonstrating that the three MoA (GR-driven cell death, CD19 signaling inhibition, and ADCC) collectively contribute to anti-tumor activity in vivo. ABBV-319 also displayed on-target depletion of normal human B-cells but did not affect peripheral NK cell counts in mice. Furthermore, CITE-seq profiling revealed that ABBV-319 treatment of human PBMCs activated GRM-induced signature genes restricted to B-cells, demonstrating the specificity of CD19-mediated delivery of GRM.
Conclusion: ABBV-319 has potent anti-tumor activity from three distinct MoA and exhibits safety improvements compared to systemic glucocorticoids, supporting its recent progression into Phase I clinical trial.
 AACR2023 Abstract (바로가기)
AACR2023 Abstract (바로가기)
Background: Histone deacetylase 1 (HDAC1) was identified from a novel in vivo CRISPR screening platform as a target gene whose inhibition reverses α-PD1 resistance driven by loss of STK11. Histone deacetylases are a well-studied class of oncology drug targets, but existing non-isoform-selective HDAC inhibitors have few approved clinical applications due to toxicities that limit sufficient exposure in solid tumors. Our data suggest that HDAC3, an essential gene, is a primary driver of bone marrow toxicity caused by HDAC inhibitors that target multiple isoforms.
Methods: We discovered and developed TNG260, a small molecule which inhibits HDAC1 with 10-fold selectivity over HDAC3 in cells, and 500-fold selectivity for the CoREST complex over the other HDAC1-containing complexes, NuRD and Sin3. Due to its CoREST-selective deacetylase inhibition, we have termed TNG260 a CoreDAC inhibitor.
Results: Treatment of an α-PD1 resistant STK11-mutant MC38 syngeneic tumor model with TNG260 re-sensitizes this model to treatment with α-PD1. The combination of TNG260 and α-PD1 led to durable complete tumor regressions in the majority of treated animals. All mice with complete responses remained tumor-free until tumor rechallenge (21 days) and rejected engraftment of tumor cells. Unlike previously developed HDAC inhibitors designed for tumor cell cytotoxicity, TNG260 has no anti-tumor efficacy in immunocompromised mice, indicating the tumor cell killing with TNG260 is immune-mediated and not due to direct cell killing. Immune profiling of tumors following treatment with TNG260 and α-PD1 showed a decoupling of Teffector and Tregulatory cell recruitment caused by α-PD1 monotherapy, leading to a more active immune microenvironment. TNG260 also decreased intratumoral infiltration of neutrophils, an immune suppressive cell type associated with STK11-mutant NSCLC. Toxicity profiling of TNG260 shows it has less viability impact on erythroid and myeloid cells in vitro than other HDAC inhibitors, and in vivo toxicity studies showed bone marrow suppression only at TNG260 doses that are no longer selective for HDAC1/2.
Conclusions: TNG260 is a potent, highly selective small molecule CoreDAC inhibitor with good drug-like properties. It reverses the immune evasion phenotype caused by loss of STK11 and induces tumor regressions in an STK11-mutant model in combination with α-PD1. The TNG260 clinical development plan will be among the first to combine the power of genetic patient selection and immunotherapy, evaluating patients with STK11 mutant cancers in a trial combining TNG260 and a checkpoint inhibitor.
TNG260 발표자료 (바로가기)
CoREST Program (바로가기)
 AACR2023 Abstract (바로가기)
AACR2023 Abstract (바로가기)
Development of MRT-2359, an orally bioavailable GSPT1 molecular glue degrader, for the treatment of lung cancers with MYC-induced translational addiction
MYC transcription factors are well-established drivers of human cancers but despite being amongst the most frequently altered oncogenes, no approved therapy targeting MYC-driven tumors has been developed to date. MYC-driven cancers are known to be addicted to protein translation. This addiction creates a dependency on critical components of the translational machinery providing in turn a unique opportunity for therapeutic intervention. We hypothesized that targeting the translation termination factor GSPT1, a key regulator of protein synthesis, would constitute a vulnerability for MYC-driven tumors. Herein we further describe MRT-2359 a potent, selective and orally bioavailable degrader of GSPT1. MRT-2359 was rationally designed using our QuEENTM discovery engine and optimized to achieve a profound and preferential antiproliferative activity in MYC-driven cell lines, such as high N- and L-MYC mRNA expressing non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC) lines. In line with expectations, MRT-2359 activity is dependent on both CRBN and the GSPT1 G-loop degron. We further demonstrate using an inducible system that the sole expression of either N- or L-MYC is sufficient to sensitize initially resistant NSCLC cells to MRT-2359. These studies therefore establish a causal link between N- and L-MYC expression and sensitivity to MRT-2359. Unlike MRT-2359, agents targeting the protein translation initiation machinery or repressing MYC transcription (CDK9 inhibitor) failed to show such differential activity. Mechanistically, RiboSeq and polysome profiling revealed that treatment with MRT-2359 in the N- or L-MYC high cell lines induces ribosome stalling at the stop codon, increased monosomes and decreased polysomes. These changes are indicative of translational repression and were confirmed using puromycilation assays. Proteomics and RNAseq studies finally demonstrated a significant reduction in the total levels of N- or L-MYC leading in turn to the downmodulation of MYC target genes. Despite robust degradation of GSPT1, no marked effect was observed in these assays in low N- or L-MYC lines, confirming the selective activity of MRT-2359 in MYC-driven lung cancers. Last, the anti-tumor activity of MRT-2359 was assessed in >80 lung patient-derived xenografts (PDXs). MRT-2359 demonstrated preferential activity in N- and L-MYC high NSCLC and SCLC PDXs, including numerous instances of tumor regressions, when dosed orally daily or intermittently. Similar levels of anti-tumor activity were also observed in neuroendocrine lung cancer and lymphoma PDXs. Together these results warrant further investigations in the clinic. Oral MRT-2359 is currently in a Phase 1/2 clinical trial in selected cancer patients with MYC-driven NSCLC, SCLC, high grade neuroendocrine cancers and diffuse large B-cell lymphoma (NCT05546268).
 AACR2023 abstract (바로가기)
AACR2023 abstract (바로가기)
BI 907828: A highly potent MDM2-p53 antagonist suitable for intermittent dose schedules
The E3 ligase MDM2 controls the tumor suppressor function of p53, which is encoded by the TP53gene. Compounds designed to bind to MDM2 preventing its interaction with p53 restore the ability of p53 to combat the pathogenic processes that underly cancer development. It has become clear from early clinical experience that thrombocytopenia represents an on-target, dose-limiting toxicity that may restrict the therapeutic utility of first-generation MDM2 inhibitors. Dosing less frequently, while maintaining efficacious exposure levels, has been proposed as an approach to mitigate side effects and improve the therapeutic window. In this -Late Breaking- session we present the discovery and preclinical evaluation of BI 907828, a highly potent, orally bioavailable and selective molecule binding to MDM2 and preventing its interaction with p53. The compound was designed to have a long half-life to enable intermittent dosing schedules in the clinic where it showed a manageable safety profile in a Phase Ia/Ib, dose-escalation/expansion study and encouraging signs of antitumor activity in patients with advanced DDLPS and WDLPS. BI 907828 is actively investigated in several clinical trials including the Phase II/III Brightline-1 study that aims to evaluate whether it is superior to doxorubicin in the first-line treatment of advanced/metastatic DDLPS. BI 907828 belongs to the class of spiro-oxindole MDM2-p53 antagonists and was discovered starting from an optimized core structure with improved chemical stability. Structure-based medicinal chemistry optimization including rigidification of the scaffold were key to accomplish the desired profile which was tested preclinically in several in vivo MDM2-amplified xenograft models where treatment with BI 907828 showed efficient tumor growth inhibition and regression.
논문발표내용 (바로가기)
Journal of Clinical Oncology 41, no. 4_suppl (February 01, 2023) 543-543.
Background: Standard-of-care treatment for advanced biliary tract cancer (BTC) is chemotherapy and outcomes remain poor; hence, there is a clinical need for effective targeted treatments. As MDM2 is a negative regulator of the tumor suppressor p53, blocking the MDM2–p53 interaction is a potential antitumor strategy. Further, preliminary data indicate that MDM2 amplification is a negative prognostic marker in BTC. The MDM2–p53 antagonist BI 907828 has shown preclinical antitumor activity in a range of malignancies and is currently being assessed in two phase Ia/Ib dose-escalation/expansion trials in patients with advanced solid tumors: as monotherapy (NCT03449381) and in combination with an anti-PD-1 antibody, ezabenlimab (NCT03964233). Here, we present data for patients with advanced BTCs in these trials.
Methods: Patients in the monotherapy trial received escalating doses of BI 907828 on day 1 of 21-day cycles (q3w). Patients in the combination trial received escalating doses of BI 907828 and 240 mg ezabenlimab q3w (doublet); one patient also received the anti-LAG-3 antibody BI 754111 (a drug that has since been discontinued; triplet).
Results: A total of 8 patients with BTC were enrolled in the 2 trials, 4 in the monotherapy trial and 4 in the combination trial. In the monotherapy trial, 2 patients had ampullary carcinoma (both received BI 907828 45 mg q3w), and 2 had cholangiocarcinoma (CC; 1 received BI 907828 45 mg q3w and 1 with intrahepatic CC [iCC] received 80 mg q3w). In the combination trial, 3 patients with iCC received 30 mg/45 mg BI 907828 doublet or 45 mg triplet, and 1 with gallbladder carcinoma (GBC) received BI 907828 45 mg doublet. Across both trials, 5 patients achieved PR, 2/4 in the monotherapy trial and 3/4 in the combination trial. In the monotherapy trial, the responding patients had iCC (80 mg q3w; MDM2-amplified; 73% tumor shrinkage; PFS event at 404 days) and ampullary adenocarcinoma (45 mg q3w; MDM2-amplified; 51% tumor shrinkage; ongoing, PFS censored at 255 days). In the combination trial, all 3 responding patients had MDM2-amplified biliary tract adenocarcinoma (2 iCC, 1 GBC); tumor shrinkage was 49–54%; PFS was 162–241 days. A further 2 patients (1 in each trial) achieved stable disease (SD). In the monotherapy trial, most common grade ≥3 treatment-related AEs (TRAEs) were thrombocytopenia, decreased white blood cell (WBC) count and neutropenia (2 patients each). In the combination trial, most common grade ≥3 TRAEs were anemia and decreased WBC count (3 patients each), neutropenia and thrombocytopenia (2 patients each).
Conclusions: The MDM2–p53 antagonist BI 907828 has shown a manageable safety profile and encouraging preliminary efficacy in patients with BTC, with 5 PRs and 2 SD in 8 patients. A phase IIa/IIb trial of BI 907828 (45 mg q3w) in patients with BTC is planned to start recruitment at the end of 2022 (NCT05512377). Clinical trial information: NCT03449381 and NCT03964233
 AACR2023 Abstract (바로가기)
AACR2023 Abstract (바로가기)
Therapeutic potential of EO-3021/SYSA1801, a Claudin18.2 antibody-drug conjugate, for the treatment of CLDN18.2-expressing cancers
Background: Claudin18.2 (CLDN18.2), a tight junction protein normally expressed only on gastric mucosa, is overexpressed in gastric, pancreatic, esophageal, ovarian, lung and other solid tumors. Unlike in normal tissue, CLDN18.2 is exposed on epithelial surfaces in malignancy. There are no approved targeted therapies for CLDN18.2-expressing cancers. An antibody-drug conjugate (ADC) composed of a monoclonal antibody (mAb) targeting CLDN18.2 with a monomethyl auristatin E (MMAE) payload site-specifically conjugated via a cleavable linker at a drug-antibody ratio (DAR) of 2, EO-3021/SYSA1801, was developed to target CLDN18.2-expressing cancer cells, minimize toxicities and maximize therapeutic index.
Methods: HEK293-CLDN18.2 cells were used to evaluate CLDN18.2-dependent EO-3021 endocytosis, MMAE payload release, inhibition of proliferation and antibody dependent cellular cytotoxicity (ADCC). Cell cycle distribution and caspase-3/7 activity were measured 24 hours after treating BxPC3-CLDN18.2 pancreatic cells with EO-3021 or the unconjugated antibody, EO-3021 mAb. Inhibition of proliferation by EO-3021 was measured on cell lines ectopically expressing medium to high CLDN18.2 and on cell lines with endogenous low to medium CLDN18.2 expression. Xenograft studies evaluating tumor growth inhibition by EO-3021, EO3021 mAb, and cisplatin or gemcitabine were done in gastric (NUGC4; NUGC4-CLDN18.2), pancreatic (Patu8988S; BxPC3-CLDN18.2), and lung (NCI-H460) cancer models.
Results: EO-3021 binding to cancer cells, endocytosis, MMAE release, and inhibition of proliferation were dependent on CLDN18.2 expression. EO-3021 (EC50: 172 ng/ml) and EO-3021 mAb (EC50: 130 ng/ml) demonstrated similar levels of ADCC. EO-3021 but not EO-3021 mAb promoted G2/M cell cycle arrest and apoptosis and exhibited potent activity across cell lines with low, medium, and high CLDN18.2 expression (IC50: 7-456 ng/mL). EO-3021 induced tumor regressions with a single dose across low, medium, and high CLDN18.2-expressing in vivo models derived from pancreatic (2-10 mg/kg), gastric (0.5-10 mg/kg), and lung cancers (4 mg/kg), respectively. In contrast, standard of care (SOC) chemotherapies and EO-3021 mAb did not induce tumor regressions across in vivo models.
Conclusions: EO-3021 demonstrated profound in vivo antitumor activity and outperformed SOC in gastric, pancreatic, and lung cancer models. Results from in vitro and in vivo studies highlight the promising therapeutic potential of EO-3021/SYSA1801 for patients with CLDN18.2-expressing cancers. A Phase 1 study is ongoing to evaluate the safety, tolerability, pharmacokinetics and preliminary anti-tumor activity of SYSA1801 in patients with CLDN18.2 positive advanced solid tumors (NCT05009966).
참고자료 (바로가기)
Drug Hunter : AACR Orlando 2023: New Drug Candidates
(Reviewed by Julien Lefranc, PhD, Edited by Dennis X. Hu, PhD.)
| (1) RMC-6291: A tri-complex KRAS-G12C(ON) inhibitor (Revolution Medicines Inc.) (2) ARV-766: An androgen receptor degrading PROTAC for mCRPC (Arvinas) (3) BAY 2965501: A first-in-class a diacylglycerol kinase zeta (lipid kinase) inhibitor for cancer immunotherapy (Bayer/Nuvisan) (4) CFT1946: A CRBN-based oral mutant-selective BRAFV600X degrader (C4 Therapeutics) (5) FX-909: A first-in-class covalent inverse agonist of the PPARG lineage transcription factor (Flare Therapeutics) (6) ABBV-319: A first-in-class glucocorticoid receptor modulator agonist ADC for B-cell malignancies (AbbVie, SSF) (7) TNG260: An HDAC1/LSD1/RCOR1 (CoREST) deacetylase complex inhibitor (Tango Therapeutics) (8) MRT-2359: A CRBN-based GSPT1 degrader (Monte Rosa Tx) (9) BI 907828: An MDM2-p53 protein-protein interaction antagonist related to the Nutlin series (Boehringer Ingelheim) (10) EO-3021: A MMAE-based CLDN18.2 ADC (Elevation Oncology) |
(1) RMC-6291: A tri-complex KRAS-G12C(ON) inhibitor (Revolution Medicines Inc.)
Discovery of RMC-6291, a tri-complex KRAS-G12C(ON) inhibitor대부분의 기존 KRAS 저해제는 inactive, GDP-bound form of KRAS와 결합하여 작용하는 반면, ‘RMC-6291’은 activated, GTP-bound forms of KRAS와 공유결합을 통해서 비가역적으로(irreversible) 지속적으로(durable) KRAS-G12C-mutated tumors에 효과를 나타낸다.
AACR2023 Abstract (바로가기)
The KRAS-G12C mutation occurs in 11 - 14% of non-small cell lung cancers, ~4% of colorectal cancers, and ~2% of pancreatic cancers in the U.S., and drives these cancers by shifting the cellular equilibrium of KRAS towards the GTP-bound, active state, KRAS-G12C(ON). The resulting increased levels of KRAS-G12C(ON) in turn increase signaling output to initiate and support the oncogenic state. RMC-6291 is a potent, orally bioavailable inhibitor of KRAS-G12C(ON) that forms a tri-complex within tumor cells along with KRAS-G12C(ON) and cyclophilin A (CypA), driving a near-immediate disruption of RAS effector binding and extinction of KRAS-G12C(ON) signaling. RMC-6291 treatment produces deep and durable suppression of RAS pathway activity in KRAS-G12C tumor models and drives profound tumor regressions in vivo. In a mouse clinical trial with a representative panel of xenograft models of KRAS-G12C NSCLC, RMC-6291 outperformed adagrasib, displaying a potential ‘best-in-class’ profile with an increased number of responses, greater depth of tumor regressions and improved durability of responses. Thus, RMC-6291 is a clinical stage inhibitor of KRAS-G12C(ON) that potentially overcomes limitations of first-generation KRAS-G12C(OFF) inhibitors.
(2) ARV-766: An androgen receptor degrading PROTAC for mCRPC (Arvinas)
Discovery of ARV-766, an androgen receptor degrading PROTAC® for the treatment of men with metastatic castration resistant prostate cancer (mCRPC)AACR2023 Abstract (바로가기)
Prostate cancer is the second leading cause of cancer death in men in the United States. The androgen receptor (AR) plays critical roles in both early disease and advanced prostate cancer. Current therapeutic approaches targeting the androgen/AR axis are initially effective, but castration resistant prostate cancer (CRPC) inevitably develops. CRPC is linked with increased AR activity via gene overexpression, amplification, and gain-of-function mutations. To address these mechanisms of AR-dependent prostate tumor growth, we have developed a novel therapeutic agent, ARV-766, a proteolysis targeting chimera (PROTAC®) that induces a protein-protein interaction between the AR and specific E3 ubiquitin ligase complexes, leading to the ubiquitination of AR and its subsequent degradation via the proteasome. In vitro, ARV-766 degrades AR in various prostate cancer cell lines, including those harboring resistance-conferring, clinically relevant point mutations, with a half-maximal degradation concentration (DC50) of <1 nM in wild type VCaP. Importantly ARV-766 also maintains potency against the AR L702H mutant, which has been associated with resistance to some AR antagonists. In vivo, ARV-766 is orally bioavailable and robustly degrades AR with a >90% observed maximum degradation (Dmax) at efficacious doses. ARV-766 significantly and dose-dependently inhibits tumor growth in murine LNCaP and VCaP xenograft models, including an enzalutamide-insensitive non-castrated VCaP model. These preclinical data supported the clinical development of ARV-766 for the treatment of men with metastatic CRPC. Selected pre-clinical data along with the chemical structure of ARV-766 will be presented.
(3) BAY 2965501: A first-in-class a diacylglycerol kinase zeta (lipid kinase) inhibitor for cancer immunotherapy, (Bayer/Nuvisan)

 BAY 2965501: DGK-zeta Lipid Kinase Inhibitor(바로가기)
BAY 2965501: DGK-zeta Lipid Kinase Inhibitor(바로가기)
In the New Drugs on the Horizon session at AACR, Bayer presented preclinical data on its novel selective diacylglycerol kinase (DGK) zeta inhibitor BAY 2965501, which is under Phase 1 clinical evaluation. The enzyme DGKzeta is a lipid kinase that can down-modulate T-cell activation by catalyzing the conversion of diacylglycerol to phosphatidic acid, thus acting as a ligand-independent, intracellular immune checkpoint. An inhibition of DGKzeta has been demonstrated to enhance T-cell priming against suboptimal tumor antigens and has the potential to overcome multiple immune-suppressive mechanisms in the tumor microenvironment. BAY-2965501 is under development for the treatment of skin cancer, kidney cancer, stomach cancer, solid tumor, gastroesophageal junction adenocarcinoma and non-small cell lung cancer. The therapeutic candidate administered through oral route and acts by targeting diacylglycerol kinase zeta (DGKZ).(4) CFT1946: A CRBN-based oral mutant-selective BRAFV600X degrader (C4 Therapeutics)
The discovery and characterization of CFT1946: A potent, selective, and orally bioavailable degrader of mutant BRAF for the treatment of BRAF-driven cancersAACR2023 Abstract (바로가기)
The BRAF kinase plays a critical role in the MAPK signaling pathway and is mutated in ~8% of all human cancers including melanoma (~60%), thyroid (~60%), and lung adenocarcinoma (~10%). The most common mutation in BRAF is V600E (Class I), which is found in >70% in these cancers. Despite the clinical success of approved small molecule inhibitors of BRAF V600E (vemurafenib, dabrafenib and encorafenib), this remains an area of unmet medical need because nearly all patients progress, due to either primary or acquired resistance. A bifunctional degradation activating compound (or BiDACTM degrader) may address the liabilities of approved drugs by overcoming, or preventing the emergence of, resistance to BRAF inhibitors. Here we describe CFT1946, an orally bioavailable cereblon-based BiDAC degrader of BRAF V600 mutant proteins, and provide an overview of the medicinal chemistry path leading to its discovery. CFT1946 degrades BRAF V600 mutant proteins, while maintaining exquisite selectivity against the proteome, sparing wild type BRAF (BRAF-WT), ARAF, and CRAF. In A375 cells, CFT1946 potently degraded BRAF V600E and inhibited ERK phosphorylation and cell growth while having no effect in the mutant KRAS, BRAF-WT driven cell line HCT116. In the A375 xenograft model, oral delivery of CFT1946 at 10 mg/kg PO BID resulted in deeper and more durable tumor regression compared to a clinically relevant dose of encorafenib. Further evaluation of CFT1946 in an engineered, clinically relevant BRAFi-resistant A375 cell line (endogenous BRAF V600E + engineered expression of NRAS Q61K) demonstrated that CFT1946 both degraded BRAF V600E and caused a loss of viability in these cells, while treatment with encorafenib had no effect. In xenografts derived from this BRAFi-resistant cell line, oral dosing of CFT1946 as a single agent led to tumor growth inhibition, while treatment with a clinically relevant dose of encorafenib had no effect on tumor growth. Furthermore, dosing CFT1946 in combination with the MEK inhibitor, trametinib, resulted in significant tumor regression, whereas combining encorafenib with the same dose of trametinib had no effect. The medicinal chemistry campaign resulting in CFT1946 focused on the improvement of in vivo pharmacokinetics and rational linker design to achieve high oral bioavailability in a beyond Rule of 5 heterobifunctional degrader. The preclinical data presented herein support the planned Phase 1/2 clinical trial of CFT1946 for the treatment of BRAF-V600 mutant solid tumors.
(5) FX-909: A first-in-class covalent inverse agonist of the PPAR gamma lineage transcription factor (Flare Therapeutics)
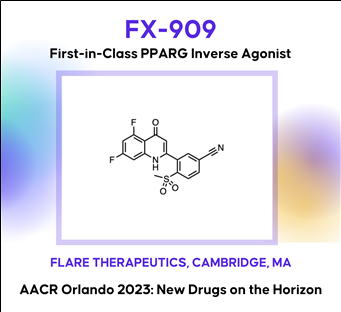 Discovery of FX-909, a first-in-class inverse agonist of the peroxisome proliferator-activated receptor gamma (PPARG) lineage transcription factor, to potentially treat patients with the luminal subtype of advanced urothelial cancer (UC)
Discovery of FX-909, a first-in-class inverse agonist of the peroxisome proliferator-activated receptor gamma (PPARG) lineage transcription factor, to potentially treat patients with the luminal subtype of advanced urothelial cancer (UC)
AACR2023 Abstract (바로가기)Although the treatment landscape of advanced urothelial cancer (UC) has changed dynamically over the last decade, metastatic UC still has amongst the worst 5-year recurrence rate of any cancer type, highlighting the need for new therapeutic options for these patients. Analogous to breast cancer, advanced UC is a heterogeneous disease, with luminal and basal subtypes. For luminal breast cancer, small molecule targeting of the lineage-defining transcription factor estrogen receptor (ER) is a demonstrated and effective therapeutic strategy. Two-thirds of advanced UC is classified as luminal, which is characterized by overexpression of the urothelial lineage-defining transcription factor PPARG. Thus, small molecule targeting of PPARG in luminal UC may serve as a therapeutic strategy analogous to ER in luminal breast cancer. While PPARG agonists have been well studied in the context of metabolic disease, PPARG inhibitors have received far less attention. Previously reported PPARG inhibitors display minimal phenotypic activity in pre-clinical UC models, calling into question the performance of these molecules, or alternatively, the role of PPARG as a survival lineage oncogene in UC. Leveraging reconstituted, multi-component PPARG biochemical systems, we first identified the limitations of previous tool compounds and then discovered a novel, covalent lead series with unique, context-dependent electrophilic properties. These efforts lead to the discovery of FX-909, a first-in-class covalent PPARG inverse agonist with powerful repressive conformational biasing activity. FX-909 is highly potent in cells (IC50=1 nM), demonstrates high specificity for PPARG (>2000-fold selective over PPARA/PPARD), and elicits robust in vitro growth inhibition in UC cell lines with activated PPARG signaling. Tumor regression was observed in UMUC9 (PPARG amplification) and HT1197 (RXRAS427F hotspot mutation) xenograft models of UC with oral dosing at 1 mg/kg. Predictable, on-target and reversible pharmacology was observed at FX-909 doses above 1 mg/kg, mimicking PPARG loss-of-function mutations with notable tissue remodeling in adipose tissue and the normal urothelium. These collective findings corroborate the role of PPARG as a key UC survival oncogene and suggest that FX-909 will be an effective therapy for patients with advanced UC harboring the luminal subtype.
(6) ABBV-319: A first-in-class glucocorticoid receptor modulator agonist ADC for B-cell malignancies (AbbVie, SSF)
Preclinical development of ABBV-319: a CD19-targeting glucocorticoid receptor modulator (GRM) agonist antibody-drug conjugate (ADC) for the treatment of B-cell malignancies
AACR2023 Abstract (초록바로가기)
Introduction: Glucocorticoids are key components of standard-of-care treatment regimens (e.g., R-CHOP, Hyper-CVAD) for several B-cell malignancies. However, prolonged systemic glucocorticoid treatment results in glucocorticoid-associated adverse events and acquired resistance that limit its therapeutic potential. ABBV-319 is a novel CD19-targeting ADC engineered to reduce glucocorticoid-associated toxicity observed with systemic glucocorticoids while possessing three distinct mechanisms of action (MoA) to increase efficacy: 1) antibody-mediated delivery of GRM to activate glucocorticoid receptor (GR) induced cell death in cancer cells, 2) inhibition of CD19 signaling, and 3) enhanced Fc-mediated cancer cell killing via afucosylation of the antibody backbone.
Results: We identified a GRM agonist that is 15 and 150 times more potent at driving GR transcriptional activation and cell death compared to clinical glucocorticoids dexamethasone and prednisolone, respectively. The conjugation of GRM agonist as the payload on ABBV-319 enables potent GRM-driven anti-cancer activity against malignant B-cell lines in vitro as well as in cell-line and patient derived xenograft (CDX and PDX) models in vivo. Remarkably, a single-dose of ABBV-319 induced sustained tumor regression and enhanced anti-tumor activity compared to repeat dosing of systemic glucocorticoids (e.g., prednisolone) at its maximum tolerated dose in mice. The CD19 monoclonal antibody (mAb) also reduced proliferation of a subset of B-cell malignant cell lines through inhibition of the PI3K/AKT pathway. Moreover, afucosylation of the CD19 mAb in ABBV-319 enhanced Fc-mediated antibody-dependent cellular cytotoxicity (ADCC), and this activity was maintained after conjugation with GRM payloads. ABBV-319 bound similarly to both V158 (high-affinity) and F158 (low-affinity) FcγRIIIa allotypes and mediated potent ADCC in co-culture assays with human peripheral blood mononuclear cells (PBMCs). Notably, ABBV-319 displayed superior efficacy compared to afucosylated CD19 mAb in human CD34+ hematopoietic stem cell-engrafted NSG-Tg(Hu-IL15) mice, demonstrating that the three MoA (GR-driven cell death, CD19 signaling inhibition, and ADCC) collectively contribute to anti-tumor activity in vivo. ABBV-319 also displayed on-target depletion of normal human B-cells but did not affect peripheral NK cell counts in mice. Furthermore, CITE-seq profiling revealed that ABBV-319 treatment of human PBMCs activated GRM-induced signature genes restricted to B-cells, demonstrating the specificity of CD19-mediated delivery of GRM.
Conclusion: ABBV-319 has potent anti-tumor activity from three distinct MoA and exhibits safety improvements compared to systemic glucocorticoids, supporting its recent progression into Phase I clinical trial.
(7) TNG260: An HDAC1/LSD1/RCOR1 (CoREST) deacetylase complex inhibitor (Tango Therapeutics)
AACR2023 Abstract (바로가기)Background: Histone deacetylase 1 (HDAC1) was identified from a novel in vivo CRISPR screening platform as a target gene whose inhibition reverses α-PD1 resistance driven by loss of STK11. Histone deacetylases are a well-studied class of oncology drug targets, but existing non-isoform-selective HDAC inhibitors have few approved clinical applications due to toxicities that limit sufficient exposure in solid tumors. Our data suggest that HDAC3, an essential gene, is a primary driver of bone marrow toxicity caused by HDAC inhibitors that target multiple isoforms.
Methods: We discovered and developed TNG260, a small molecule which inhibits HDAC1 with 10-fold selectivity over HDAC3 in cells, and 500-fold selectivity for the CoREST complex over the other HDAC1-containing complexes, NuRD and Sin3. Due to its CoREST-selective deacetylase inhibition, we have termed TNG260 a CoreDAC inhibitor.
Results: Treatment of an α-PD1 resistant STK11-mutant MC38 syngeneic tumor model with TNG260 re-sensitizes this model to treatment with α-PD1. The combination of TNG260 and α-PD1 led to durable complete tumor regressions in the majority of treated animals. All mice with complete responses remained tumor-free until tumor rechallenge (21 days) and rejected engraftment of tumor cells. Unlike previously developed HDAC inhibitors designed for tumor cell cytotoxicity, TNG260 has no anti-tumor efficacy in immunocompromised mice, indicating the tumor cell killing with TNG260 is immune-mediated and not due to direct cell killing. Immune profiling of tumors following treatment with TNG260 and α-PD1 showed a decoupling of Teffector and Tregulatory cell recruitment caused by α-PD1 monotherapy, leading to a more active immune microenvironment. TNG260 also decreased intratumoral infiltration of neutrophils, an immune suppressive cell type associated with STK11-mutant NSCLC. Toxicity profiling of TNG260 shows it has less viability impact on erythroid and myeloid cells in vitro than other HDAC inhibitors, and in vivo toxicity studies showed bone marrow suppression only at TNG260 doses that are no longer selective for HDAC1/2.
Conclusions: TNG260 is a potent, highly selective small molecule CoreDAC inhibitor with good drug-like properties. It reverses the immune evasion phenotype caused by loss of STK11 and induces tumor regressions in an STK11-mutant model in combination with α-PD1. The TNG260 clinical development plan will be among the first to combine the power of genetic patient selection and immunotherapy, evaluating patients with STK11 mutant cancers in a trial combining TNG260 and a checkpoint inhibitor.
TNG260 발표자료 (바로가기)
CoREST Program (바로가기)
(8) MRT-2359: A CRBN-based GSPT1 degrader (Monte Rosa Tx)
AACR2023 Abstract (바로가기)Development of MRT-2359, an orally bioavailable GSPT1 molecular glue degrader, for the treatment of lung cancers with MYC-induced translational addiction
MYC transcription factors are well-established drivers of human cancers but despite being amongst the most frequently altered oncogenes, no approved therapy targeting MYC-driven tumors has been developed to date. MYC-driven cancers are known to be addicted to protein translation. This addiction creates a dependency on critical components of the translational machinery providing in turn a unique opportunity for therapeutic intervention. We hypothesized that targeting the translation termination factor GSPT1, a key regulator of protein synthesis, would constitute a vulnerability for MYC-driven tumors. Herein we further describe MRT-2359 a potent, selective and orally bioavailable degrader of GSPT1. MRT-2359 was rationally designed using our QuEENTM discovery engine and optimized to achieve a profound and preferential antiproliferative activity in MYC-driven cell lines, such as high N- and L-MYC mRNA expressing non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC) lines. In line with expectations, MRT-2359 activity is dependent on both CRBN and the GSPT1 G-loop degron. We further demonstrate using an inducible system that the sole expression of either N- or L-MYC is sufficient to sensitize initially resistant NSCLC cells to MRT-2359. These studies therefore establish a causal link between N- and L-MYC expression and sensitivity to MRT-2359. Unlike MRT-2359, agents targeting the protein translation initiation machinery or repressing MYC transcription (CDK9 inhibitor) failed to show such differential activity. Mechanistically, RiboSeq and polysome profiling revealed that treatment with MRT-2359 in the N- or L-MYC high cell lines induces ribosome stalling at the stop codon, increased monosomes and decreased polysomes. These changes are indicative of translational repression and were confirmed using puromycilation assays. Proteomics and RNAseq studies finally demonstrated a significant reduction in the total levels of N- or L-MYC leading in turn to the downmodulation of MYC target genes. Despite robust degradation of GSPT1, no marked effect was observed in these assays in low N- or L-MYC lines, confirming the selective activity of MRT-2359 in MYC-driven lung cancers. Last, the anti-tumor activity of MRT-2359 was assessed in >80 lung patient-derived xenografts (PDXs). MRT-2359 demonstrated preferential activity in N- and L-MYC high NSCLC and SCLC PDXs, including numerous instances of tumor regressions, when dosed orally daily or intermittently. Similar levels of anti-tumor activity were also observed in neuroendocrine lung cancer and lymphoma PDXs. Together these results warrant further investigations in the clinic. Oral MRT-2359 is currently in a Phase 1/2 clinical trial in selected cancer patients with MYC-driven NSCLC, SCLC, high grade neuroendocrine cancers and diffuse large B-cell lymphoma (NCT05546268).
(9) BI 907828: An MDM2-p53 protein-protein interaction antagonist related to the Nutlin series (Boehringer Ingelheim)
AACR2023 abstract (바로가기)BI 907828: A highly potent MDM2-p53 antagonist suitable for intermittent dose schedules
The E3 ligase MDM2 controls the tumor suppressor function of p53, which is encoded by the TP53gene. Compounds designed to bind to MDM2 preventing its interaction with p53 restore the ability of p53 to combat the pathogenic processes that underly cancer development. It has become clear from early clinical experience that thrombocytopenia represents an on-target, dose-limiting toxicity that may restrict the therapeutic utility of first-generation MDM2 inhibitors. Dosing less frequently, while maintaining efficacious exposure levels, has been proposed as an approach to mitigate side effects and improve the therapeutic window. In this -Late Breaking- session we present the discovery and preclinical evaluation of BI 907828, a highly potent, orally bioavailable and selective molecule binding to MDM2 and preventing its interaction with p53. The compound was designed to have a long half-life to enable intermittent dosing schedules in the clinic where it showed a manageable safety profile in a Phase Ia/Ib, dose-escalation/expansion study and encouraging signs of antitumor activity in patients with advanced DDLPS and WDLPS. BI 907828 is actively investigated in several clinical trials including the Phase II/III Brightline-1 study that aims to evaluate whether it is superior to doxorubicin in the first-line treatment of advanced/metastatic DDLPS. BI 907828 belongs to the class of spiro-oxindole MDM2-p53 antagonists and was discovered starting from an optimized core structure with improved chemical stability. Structure-based medicinal chemistry optimization including rigidification of the scaffold were key to accomplish the desired profile which was tested preclinically in several in vivo MDM2-amplified xenograft models where treatment with BI 907828 showed efficient tumor growth inhibition and regression.
논문발표내용 (바로가기)
Journal of Clinical Oncology 41, no. 4_suppl (February 01, 2023) 543-543.
Background: Standard-of-care treatment for advanced biliary tract cancer (BTC) is chemotherapy and outcomes remain poor; hence, there is a clinical need for effective targeted treatments. As MDM2 is a negative regulator of the tumor suppressor p53, blocking the MDM2–p53 interaction is a potential antitumor strategy. Further, preliminary data indicate that MDM2 amplification is a negative prognostic marker in BTC. The MDM2–p53 antagonist BI 907828 has shown preclinical antitumor activity in a range of malignancies and is currently being assessed in two phase Ia/Ib dose-escalation/expansion trials in patients with advanced solid tumors: as monotherapy (NCT03449381) and in combination with an anti-PD-1 antibody, ezabenlimab (NCT03964233). Here, we present data for patients with advanced BTCs in these trials.
Methods: Patients in the monotherapy trial received escalating doses of BI 907828 on day 1 of 21-day cycles (q3w). Patients in the combination trial received escalating doses of BI 907828 and 240 mg ezabenlimab q3w (doublet); one patient also received the anti-LAG-3 antibody BI 754111 (a drug that has since been discontinued; triplet).
Results: A total of 8 patients with BTC were enrolled in the 2 trials, 4 in the monotherapy trial and 4 in the combination trial. In the monotherapy trial, 2 patients had ampullary carcinoma (both received BI 907828 45 mg q3w), and 2 had cholangiocarcinoma (CC; 1 received BI 907828 45 mg q3w and 1 with intrahepatic CC [iCC] received 80 mg q3w). In the combination trial, 3 patients with iCC received 30 mg/45 mg BI 907828 doublet or 45 mg triplet, and 1 with gallbladder carcinoma (GBC) received BI 907828 45 mg doublet. Across both trials, 5 patients achieved PR, 2/4 in the monotherapy trial and 3/4 in the combination trial. In the monotherapy trial, the responding patients had iCC (80 mg q3w; MDM2-amplified; 73% tumor shrinkage; PFS event at 404 days) and ampullary adenocarcinoma (45 mg q3w; MDM2-amplified; 51% tumor shrinkage; ongoing, PFS censored at 255 days). In the combination trial, all 3 responding patients had MDM2-amplified biliary tract adenocarcinoma (2 iCC, 1 GBC); tumor shrinkage was 49–54%; PFS was 162–241 days. A further 2 patients (1 in each trial) achieved stable disease (SD). In the monotherapy trial, most common grade ≥3 treatment-related AEs (TRAEs) were thrombocytopenia, decreased white blood cell (WBC) count and neutropenia (2 patients each). In the combination trial, most common grade ≥3 TRAEs were anemia and decreased WBC count (3 patients each), neutropenia and thrombocytopenia (2 patients each).
Conclusions: The MDM2–p53 antagonist BI 907828 has shown a manageable safety profile and encouraging preliminary efficacy in patients with BTC, with 5 PRs and 2 SD in 8 patients. A phase IIa/IIb trial of BI 907828 (45 mg q3w) in patients with BTC is planned to start recruitment at the end of 2022 (NCT05512377). Clinical trial information: NCT03449381 and NCT03964233
(10) EO-3021: A MMAE-based CLDN18.2 ADC (Elevation Oncology)
AACR2023 Abstract (바로가기)Therapeutic potential of EO-3021/SYSA1801, a Claudin18.2 antibody-drug conjugate, for the treatment of CLDN18.2-expressing cancers
Background: Claudin18.2 (CLDN18.2), a tight junction protein normally expressed only on gastric mucosa, is overexpressed in gastric, pancreatic, esophageal, ovarian, lung and other solid tumors. Unlike in normal tissue, CLDN18.2 is exposed on epithelial surfaces in malignancy. There are no approved targeted therapies for CLDN18.2-expressing cancers. An antibody-drug conjugate (ADC) composed of a monoclonal antibody (mAb) targeting CLDN18.2 with a monomethyl auristatin E (MMAE) payload site-specifically conjugated via a cleavable linker at a drug-antibody ratio (DAR) of 2, EO-3021/SYSA1801, was developed to target CLDN18.2-expressing cancer cells, minimize toxicities and maximize therapeutic index.
Methods: HEK293-CLDN18.2 cells were used to evaluate CLDN18.2-dependent EO-3021 endocytosis, MMAE payload release, inhibition of proliferation and antibody dependent cellular cytotoxicity (ADCC). Cell cycle distribution and caspase-3/7 activity were measured 24 hours after treating BxPC3-CLDN18.2 pancreatic cells with EO-3021 or the unconjugated antibody, EO-3021 mAb. Inhibition of proliferation by EO-3021 was measured on cell lines ectopically expressing medium to high CLDN18.2 and on cell lines with endogenous low to medium CLDN18.2 expression. Xenograft studies evaluating tumor growth inhibition by EO-3021, EO3021 mAb, and cisplatin or gemcitabine were done in gastric (NUGC4; NUGC4-CLDN18.2), pancreatic (Patu8988S; BxPC3-CLDN18.2), and lung (NCI-H460) cancer models.
Results: EO-3021 binding to cancer cells, endocytosis, MMAE release, and inhibition of proliferation were dependent on CLDN18.2 expression. EO-3021 (EC50: 172 ng/ml) and EO-3021 mAb (EC50: 130 ng/ml) demonstrated similar levels of ADCC. EO-3021 but not EO-3021 mAb promoted G2/M cell cycle arrest and apoptosis and exhibited potent activity across cell lines with low, medium, and high CLDN18.2 expression (IC50: 7-456 ng/mL). EO-3021 induced tumor regressions with a single dose across low, medium, and high CLDN18.2-expressing in vivo models derived from pancreatic (2-10 mg/kg), gastric (0.5-10 mg/kg), and lung cancers (4 mg/kg), respectively. In contrast, standard of care (SOC) chemotherapies and EO-3021 mAb did not induce tumor regressions across in vivo models.
Conclusions: EO-3021 demonstrated profound in vivo antitumor activity and outperformed SOC in gastric, pancreatic, and lung cancer models. Results from in vitro and in vivo studies highlight the promising therapeutic potential of EO-3021/SYSA1801 for patients with CLDN18.2-expressing cancers. A Phase 1 study is ongoing to evaluate the safety, tolerability, pharmacokinetics and preliminary anti-tumor activity of SYSA1801 in patients with CLDN18.2 positive advanced solid tumors (NCT05009966).
편집 작성: 이현규 (한국화합물은행)
참고자료 (바로가기)
Drug Hunter : AACR Orlando 2023: New Drug Candidates
(Reviewed by Julien Lefranc, PhD, Edited by Dennis X. Hu, PhD.)